Cover Story
ADVERTISEMENT

-
August 6, 2007 - Volume 85, Number 32
- pp. 11-19
Centering On Chirality
Chemists are finding asymmetric synthesis increasingly handy for making pharmaceutical compounds at large scale
Ann M. Thayer
NATURE HAS A WAY OF KNOWING how to make things work. Reactions often run in a catalytic mode, and material use, energy, and waste are minimized. Many molecules are chiral, and their unique handedness has both intricate and dramatic influences on how they interact with biological systems.
 Courtesy of Clark Landis
Courtesy of Clark Landis
In pharmaceuticals, stereoselective interactions are profoundly important because one or the other enantiomer of a compound can have beneficial or disastrous results. Awareness of this phenomenon intensified when the teratogenic effects of thalidomide, once used as an antinausea drug for pregnant women, emerged in the 1960s. Thalidomide and many other chiral drugs had been sold for years as racemic mixtures.
In 1992, the U.S. Food & Drug Administration issued a policy on stereoisomeric drugs. Racemates can be sold???FDA still occasionally approves them, and even thalidomide is now used with appropriate warnings in chemotherapy???but the enantiomers must be characterized pharmacologically and toxicologically. Since one enantiomer could be unsafe or merely inactive baggage, and the cost of characterization is so high, the drug industry has shifted to making single-enantiomer forms of chiral compounds.
"In 2006, 80% of small-molecule drugs approved by FDA were chiral and 75% were single enantiomers," says Ian C. Lennon, scientist and technology leader for Dowpharma. Assuming no differences between chiral and achiral compounds in their discovery, attrition, or approval rates, about 200 chiral compounds could enter the development process each year.
To meet these challenges, chemists in the pharmaceutical industry are increasingly called upon to mimic what nature does so easily and so well. They may try a variety of chiral technologies, including separations, salt resolutions, and asymmetric syntheses involving chiral auxiliaries, chemocatalysis, or biocatalysis to get to a single enantiomer. Or they may start with building blocks, when available in sufficient quantity and quality at the right price, from the chiral pool.
The field of asymmetric synthesis is expanding rapidly. Academic researchers continually create new reactions or asymmetric variants of existing ones. Within industry, process chemists must adapt these to production needs to make single-enantiomer compounds practically and efficiently at large scale in extremely high purity.
Process chemists usually step in to make initial kilogram amounts of an active pharmaceutical ingredient (API) for preclinical and early clinical work. "This is the only stage during the entire drug discovery and development process where simply making the API is on the critical path between discovery and marketing a new drug," explains Karel M. J. Brands, a senior director in the process research department at Merck. "Speed is essential, and you don't always have the luxury of trying to come up with the best synthesis."
As a starting point, these chemists are frequently inspired by the medicinal chemistry route, he explains. These routes, however, are seldom practical and efficient-or environmentally benign and economically viable. Instead, they may use undesirable or hazardous reagents to make racemates, followed by chromatography to separate enantiomers.
"The deliberate design of an asymmetric synthesis comes later," Brands says, when the drug candidate is ready to move into early proof-of-concept studies. "This is when a molecule starts to become of serious interest, and we are willing to put some significant resources into coming up with the best possible synthesis."
Other routes may be developed in parallel. "Most often at Merck, the arrival of the asymmetric synthesis comes in time to answer the much bigger scale needs for the late development of an API," he says. "To have a fundamentally efficient approach to a chiral target, you have to practice asymmetric chemistry."
In the early 1990s, most chiral drugs were derived from chiral-pool materials, and only 20% of all drugs were made via purely synthetic approaches, Lennon says. "Today, just 25% come from the chiral pool and over 50% use chiral technologies."
"Asymmetric synthesis is still seen as very challenging," says Hans-J??rgen Federsel, director of science in global process R&D at AstraZeneca. "Much of it relies on catalysis, and designing a catalytic process and ensuring it operates well takes a lot of time." Outcomes are unpredictable, and because many APIs and intermediates are unprecedented as substrates, it can be difficult to even know where to begin, he adds.
 View Enlarged Image
View Enlarged Image
"Many people in the pharmaceutical industry don't want to take on that burden," Federsel says. "Why invest a lot of effort in designing a fantastic process if you will have to discontinue it a year down the line because of the compound's toxicity or lack of efficacy?" Time and cost pressures, along with the quantity of projects, often dictate that other processes will win out. "Quick methods, and preferably generic ones, are desirable to cut out as much development work as possible," he says.
"Preparative chromatography is now blooming. This was always seen as a last resort but has now been integrated into normal project work to a much higher degree," Federsel continues. "Even if it is expensive, we know that it works and that within a few days, we can produce a couple kilograms to drive a project forward."
Many company scientists say a problem with using separations for large-scale production is that the costs of making both enantiomers are incurred, but one will be discarded. Higher yields may be possible if the second enantiomer can be converted or racemized for reprocessing.
On the plus side, chiral chromatography is applicable to most chiral small molecules and is very scalable. Several drugs have been made this way at the multiton scale, including Pfizer's antidepressant Zoloft, UCB Pharma's epilepsy drug Keppra, Cephalon's insomnia drug Nuvigil, and Lundbeck's Cipralex, also marketed by Forest Laboratories as Lexapro, for depression.
"Existing commercial projects have demonstrated that it is often cheaper to produce single-enantiomer drugs with chromatography than with traditional technologies such as crystallization or asymmetric synthesis," Jean Blehaut, president of Novasep's pharma business unit, said in June when announcing a collaboration with Daicel Chemical Industries. The two companies have partnered to promote the use of chiral chromatography for producing intermediates and APIs.
Traditional methods for crystallizing diastereomeric salts have found wide use. Cipralex (escitalopram) is the S-enantiomer of the earlier racemic drug Celexa. Several resolutions have been published, a recent one using didesmethylcitalopram. Chemists at Dr. Reddy's Laboratories and collaborators in India took the somewhat unusual approach of chemically modifying a chiral amine handle on the substrate itself to improve a salt resolution (Org. Process Res. Dev. 2007, 11, 289).
Although salt resolutions require a lot of trial and error, they are usually easy to handle and simple to scale up, says Vijayavitthal T. Mathad, who moved from Dr. Reddy's to Megafine Pharma, in Mumbai, where he is vice president of R&D. "The cost of resolution processes is normally affordable, even by a small manufacturer," he adds. Solvents and inexpensive resolving agents are recoverable at high levels. Cost and time pressures may rule out exploring asymmetric synthesis, he adds, especially in the competitive generic drug market.
Scientists at Dr. Reddy's and collaborators also came up with a less expensive alternative resolution route to make an intermediate for aprepitant, the API in Merck's antiemetic drug, Emend (Org. Process Res. Dev. 2007, 11, 455). They targeted the morpholine core that has two of the drug's three chiral centers.
Another aprepitant intermediate bearing a third chiral center is (1R)-[3,5-bis(trifluoromethyl)phenyl]ethanol (BTMP). The Dr. Reddy's team enantioselectively transesterified the racemic alcohol using vinyl acetate as the acyl donor and a lipase enzyme. This resolved the unacylated Salcohol and R-acetate, which was hydrolyzed separately to the R-alcohol.
Swiss catalyst developer Solvias and its scale-up partner Novasep have published an asymmetric route they developed in just two months to produce BTMP for Merck using a ruthenium catalyst with a chiral phosphine-oxazoline ferrocenyl ligand (Org. Process Res. Dev. 2007, 11, 519). The route involves the enantioselective hydrogenation of 3,5-bis(trifluoromethyl)acetophenone.
"The enantioselective reduction of aryl ketones is an important transformation from both academic/synthetic as well as industrial points of view," they write. They ran their optimized process twice on a 140-kg scale, finding it could compete with alternatives such as transfer hydrogenation and biocatalytic and hydride reductions.
"More and more pharmaceutical companies are comfortable using asymmetric hydrogenation, although a few haven't tried it yet," Dowpharma's Lennon says. Although the technology has been around for decades, in the past five years, publications, catalysts for testing, substrates tested, suppliers, and success stories have helped it move into API synthesis.
Four drugs approved in just the past 24 months—Takeda's Rozerem (ramelteon), a sleep aid that binds to melatonin receptors; Merck's Januvia (sitagliptin), one of a new class of oral diabetes drugs; Novartis' Tekturna (aliskiren), the first hypertension drug to target the kidney enzyme renin; and Boehringer Ingelheim's Aptivus (tipranavir), an HIV protease inhibitor???have chiral centers reportedly generated via catalytic asymmetric hydrogenation.
That hydrogenation is the final synthesis step for the first two drugs is surprising, Lennon remarks. Concerns about hydrogenating valuable complex molecules and catalysts contaminating the final product made such routes almost unthinkable a few years ago (C&EN, Sept. 5, 2005, page 55).
Now, Merck makes multiple tons of the chiral β-amino acid derivative sitagliptin via the late-stage hydrogenation of an unprotected enamine, using a rhodium catalyst and the Solvias chiral ligand Josiphos. The manufacturing processes for Emend and Januvia, in large part because of their innovative use of asymmetric synthesis, won Presidential Green Chemistry Challenge Awards, Merck's Brands notes.
Merck has validated catalytic approaches many times. It got a leg up on the competition by deciding a few years ago to have a lab dedicated to discovering and developing asymmetric hydrogenations. "Within a matter of days, we can find a solid hit and systematically work our way to something that can be scaled up," Brands says.
ALONG THESE LINES, Merck Associate Director Kevin R. Campos discussed process development for taranabant at Scientific Update's Modern Synthetic Methods & Chiral USA meeting in Philadelphia last month. Now in Phase III clinical trials, the antiobesity agent blocks endocannabinoid receptors to control hunger and is a potential competitor to Sanofi-Aventis' rimonabant.
Taranabant is a hindered secondary amine with two contiguous stereocenters. In lieu of a nine-step medicinal chemistry route that used expensive and toxic materials, as well as chiral chromatography, the process group developed a dynamic kinetic resolution based on enantioselective catalytic reduction to convert a racemic ketone starting material into a single enantiomer of the corresponding alcohol in high yield (Org. Process Res. Dev. 2007, 11, 616).
By this means, they produced 65 kg of material, despite it requiring azide chemistry and work to upgrade the enantiopurity. The route also lacked solid intermediates, which limited options for production. "The chemistry is varied, and the vendors who specialize in transition-metal chemistry are not versed in handling azides and vice versa," Campos tells C&EN. "The lack of solid intermediates prevented us from transferring between vendors."
Campos and coworkers then created a route involving the stereocontrolled formation of a tetrasubstituted enamide. It relies on a Merck-developed palladium-catalyzed coupling to make the enamide (Org. Lett. 2005, 7, 1185). Rhodium-catalyzed asymmetric hydrogenation of the enamide, using a Solvias ligand, forms both chiral centers in a single step.
"We had to be innovative with the chemistry because no examples of asymmetric hydrogenation of tetrasubstituted enamides of this complexity are in the literature, especially ones that have been performed on a multikilogram scale," Campos says. "Even though the substrate is complex, the synthesis is convergent, and the chemistry is quite robust."
The route takes six steps with 54% yield and 96% enantiomeric excess (ee). Every intermediate is isolatable, and the hydrogenation product crystallizes with an upgrade to 99% ee. Because the cyanoenamide couldn't be directly hydrogenated, the process required two extra steps to hydrolyze the cyano group and then convert it back.

A Roche team recently published a new enantioselective synthesis of orlistat, the API in the antiobesity drug Xenical, designed to decrease fat absorption. Using a ruthenium-MeOBIPHEP catalyst, they hydrogenated a β-ketoester in the first step to create a critical enantiopure intermediate (Org. Process Res. Dev. 2007, 11, 524). They have produced more than two tons of the resulting β-hydroxyester at over 99% ee. The ligand is from a family of atropisomeric diphosphines invented at Roche and used in several large-scale processes.
Solvias has licensed this ligand family and makes it and other asymmetric hydrogenation catalysts available for screening and at large scale. Dowpharma makes more than 10 at kilogram scale, including Me-DuPhos, which is used in the production of several major drugs. Chiral Quest has commercialized five catalysts, some of which are being used in large-scale development projects. Other suppliers working in this area are DSM, Degussa, Takasago International, and Johnson Matthey.
Benoit Pugin, a lead scientist at Solvias, estimates that more than 3,000 chiral diphosphine ligands are known. Reasons behind the diversity are their substrate specificity and the empirical means by which they are selected and employed. Only about 5-10% of the ligands are commercially available in small quantities, and just about 1% in kilogram quantities, he says. Making larger amounts can be done by devising easy and scalable syntheses.
Along these lines, Solvias has designed new ferrocenyl ligands, including P-chiral ligands called Chenphos. This class was designed to compete with Trifer catalysts from Phoenix Chemical's Stylacats unit (Angew. Chem. Int. Ed. 2007, 46, 4141) and DSM's MonoPhos catalysts in making Novartis's Tekturna.
Among several known routes to the drug, two in production depend on catalytic asymmetric hydrogenation. A process developed by Speedel Pharmaceuticals in close collaboration with Solvias uses Solvias' Walphos. DSM scientists recently reported their mixed-ligand approach using a chiral phosphoramidite and nonchiral triarylphosphine to increase the rate and enantioselectivity (Org. Process Res. Dev. 2007, 11, 585). At the Philadelphia meeting, Pugin showed that Chenphos offers even better productivity and enantioselectivity.
WITH THE EXPANSION in chiral ligand libraries, some companies have partnered with outside specialists to introduce catalytic asymmetric processes at an early point in development. An example is Eli Lilly & Co.'s chemical product R&D group, which sought an improved route to a selective estrogen receptor β agonist for prostatic disease. At the meeting, research adviser Scott A. May discussed Lilly's efforts to develop an efficient route to a substituted benzopyran with three contiguous chiral centers.
Lilly's three-pronged assault on the stereochemistry played to its own and others' strengths. Internally, they screened classical and enzymatic resolutions. Outside providers screened metal-ligand complexes on ester and carboxylic acid versions of a challenging tetrasubstituted alkene intermediate. In the end, Lilly decided to work with an outside provider to optimize a ruthenium P-Phos complex for asymmetric hydrogenation. And Lilly conducted the pilot-plant-scale process.
In another collaboration, Dowpharma helped develop a route for Pfizer's Celsentri (maraviroc) and related backup compounds. In Phase III trials, the drug targets how HIV infects immune system cells, rather than the virus itself. U.S. and European approvals are expected this year. The collaborators succeeded in synthesizing a preferred aldehyde-enamide intermediate and asymmetrically hydrogenating it.
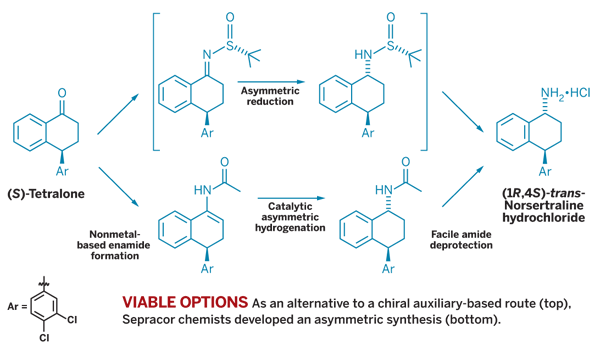
Making enamide substrates for asymmetric hydrogenation with a wide range of catalysts has become increasingly common. Scientists at Sepracor have developed large-scale stereoselective syntheses of (1R,4S)-trans-norsertraline hydrochloride, a chiral amine structurally similar to the antidepressant Zoloft but with a different pharmacological profile. One challenge was establishing two chiral centers while overcoming a substrate bias toward forming the cis diastereomer.
Their first approach, used to produce kilograms of material for clinical study, began with (S)-tetralone, which already contains one of the two needed chiral centers. Condensation with (R)-tert-butylsulfinamide (TBSA), which acts as a chiral directing group to dictate stereochemistry, leads to a sulfinyl imine that can be stereoselectively reduced and then hydrolyzed to the product (Org. Process Res. Dev. 2007, 11, 726).
"This synthesis was very quick to develop and scale up, which is why chiral reagents or resolutions are among the first processes we lean toward," says Roger P. Bakale, Sepracor's executive director for chemical process R&D.
Looking for an alternative to using expensive (R)-TBSA, a Sepracor team began scouting for other routes starting with the benzylic ketone tetralone. "An obvious approach is the asymmetric hydrogenation of an enamide, but there are not many published procedures for making enamides from benzylic ketones," explains research fellow Surendra P. Singh.
Instead, Singh and coworkers tackled a novel non-metal-based route to the enamide. After succeeding, they worked with Dowpharma and found an efficient Norphos rhodium catalyst for the hydrogenation. They then came up with a convenient process to deprotect the amide without racemization and yield the amine.
"We've scaled it up to make more than 50 kg of the API," Singh says. Details will appear in patents and publications. For now, Singh says, the result is a short and scalable process with an overall yield of 63% from the ketone. Besides the novel chemistries, it uses an unpatented catalyst to give the product in 97% diastereomeric excess (de) and 99.9% de after crystallization.

Singh is proud that a smaller drug company team with limited resources created the norsertraline process from scratch. "Not that many asymmetric syntheses are used at large scale," he points out, "and we feel good that we could do it."
Although catalyst producers and many process chemists consider hydrogenation to be among the more mature methods in asymmetric synthesis, it's not yet routine. And, Dowpharma's Lennon cautions, "asymmetric hydrogenation doesn't make a process by itself. It's effective only if the whole process is effective."
Hydrogenations of functionalized olefins and ketones are probably the easier asymmetric transformations to do, Merck's Brands explains, whereas catalytic asymmetric carbon-carbon bond formation in a truly economic fashion is much more difficult. New synthetic approaches will gradually be adopted and should become apparent as drug candidates advance. At the same time, because attrition is so high, many processes will never be demonstrated on a manufacturing scale.
"Outside of hydrogenation, no other form of asymmetric catalysis is being broadly applied at large scale," Lennon adds. Dowpharma is optimistic that hydroformylation will be next, and it is developing achiral and chiral ligands. The atom-efficient reaction combines an olefin, hydrogen, and carbon monoxide to form an aldehyde, which is a useful synthetic building block.
THE COMPANY has worked with University of Wisconsin, Madison, chemistry professor Clark R. Landis, who has developed stable and active diazaphospholane ligands for enantioselective hydroformylation. The ligands can be easily synthesized from inexpensive starting materials and resolved to give both useful antipodes. They tolerate a range of functional groups to alter steric and electronic properties and offer practical enantioselectivities for a wide variety of substrates.
Asymmetric hydroformylation can present challenges around chemo-, enantio-, and regioselectivity, as well as catalyst stability and reactivity. But the situation is improving. "There are fewer technical hurdles than business ones," says Robert B. Appell, technical sales and process R&D leader at Dowpharma. "Companies don't want to be beta testers and would like to see large-scale examples before they buy in." He says some are showing interest in catalyst screening.
Hydroformylation requires different retrosynthetic thinking about where aldehydes might serve as intermediates in synthetic routes, points out Xumu Zhang, who founded catalyst producer Chiral Quest in 2000. He is also a chemistry professor at Rutgers, the State University of New Jersey, where he will head a new center for molecular catalysis.
In 2006, Zhang and graduate student Yongjun Yan designed a phosphine-phosphite ligand for asymmetric hydroformylation called YanPhos (J. Am. Chem. Soc. 2006, 128, 7198). More recently, Zhang and coworkers have made diphosphite ligands with binaphthyl backbones (Tetrahedron Lett. 2007, 48, 4781).
Recent results have shown that catalysts designed for hydrogenation work well in hydroformylation and vice versa, as well as in other transformations. Besides transition-metal-based asymmetric catalysis, industrial researchers employ a variety of synthetic strategies, including reactions with chiral directing groups or auxiliaries, organocatalysts, and biocatalysts (C&EN, Aug. 14, 2006, page 15).
The usefulness of the aminoindanol moiety as a chiral auxiliary was highlighted about 15 years ago in Merck's synthesis of the HIV drug Crixivan, which at the time was one of the most complex molecules the company's chemists had synthesized (C&EN, June 20, 2005, page 54). Among those researchers was Chris H. Senanayake, now Boehringer Ingelheim's vice president for chemical development.
"In general, rapid development of enantiopure APIs is a highly challenging task, and chiral auxiliary-based approaches are still a frequently employed strategy," Senanayake says. Notable advantages include the ready availability of some powerful chiral auxiliaries as well as the proven generality with respect to substrates. In addition, these approaches are often easy to scale up in a timely manner.
Boehringer Ingelheim's process chemists have used different auxiliary-based routes to make intermediates containing trifluoromethyl-substituted quaternary stereogenic centers. "The structural motif of the tertiary alcohol with a CF3 group attached to the carbon center is very difficult to install, and new methodologies needed to be developed," says Boehringer Ingelheim senior principal scientist Jinhua Jeff Song.
In one case, the process involved generating a p-tolyl methyl sulfoxide auxiliary in situ and then reacting it with a trifluoromethyl ketone to form the quaternary center in a one-pot operation (Org. Process Res. Dev. 2007, 11, 605). Meanwhile, Song and coworkers discovered a novel direct asymmetric trifluoromethylation of a keto ester using a trans-2-phenylcyclohexanol auxiliary (J. Org. Chem. 2007, 72, 292) that provided the CF3-substituted product with much higher selectivity.
"But the most practical and cost-efficient approach used an asymmetric acetate aldol reaction directed by an aminoindanol-derived chiral auxiliary," Song explains. After preparing chiral acetate with the aminoindanol in a one-pot process, they formed a lithium enolate and reacted it with a trifluoromethyl ketone to obtain the aldol product. Then, after crystallizing the major diastereomer, they cleaved off the auxiliary by transesterification and recovered it (Org. Process Res. Dev. 2007, 11, 534).
Likewise, GlaxoSmithKline researchers published three generations of syntheses for making a tetrahydrocarbazole compound as a potential treatment for human papillomavirus infections, considered the most common sexually transmitted disease (Org. Process Res. Dev. 2007, 11, 539).
Progressing from an initial racemic synthesis using chromatography or salt resolution, their next best route was a modestly successful enantioselective reductive amination via chiral transfer hydrogenation with Noyori-type ruthenium catalysts. The most efficient and selective synthesis was a diastereoselective reductive amination directed by a chiral phenylethylamine auxiliary.
"Enantioselective synthesis, if achieved in satisfactory selectivity, would be more desirable," GSK research investigator Shiping Xie says. That's because the auxiliary was required in stoichiometric quantities and had to be attached and removed. "These are clearly disadvantages, but fortunately the auxiliary was not expensive," Xie says. Better yet, the reactions worked well and were scaled up to provide multikilograms of the compound at over 99.5% ee.
GSK scientists also reported an optimized asymmetric phase-transfer catalyzed (PTC) alkylation to make a nonnatural amino acid intermediate for a drug in development (Org. Process Res. Dev. 2007, 11, 624). PTC reaction conditions typically are mild and use environmentally benign reagents and solvents. Their process used a cinchona alkaloid-derived chiral catalyst and avoided stoichiometric amounts of chiral auxiliaries and azide chemistry used in an earlier route. They believe it is one of very few published examples of asymmetric PTC using secondary alkyl halides.
Research adviser Nicholas A. Magnus and coworkers at Lilly recently developed what they believe is the largest-scale demonstration of an enantioselective aryl transfer reaction. The goal was to produce a chiral diarylmethanol intermediate that could be converted to an acetate and subsequently used to make kilograms of compounds for development as mGlu2 receptor potentiators for treating migraine headaches (Org. Process Res. Dev. 2007, 11, 560).
"We considered all the typical known methods for converting an appropriate ketone into a chiral secondary alcohol, including asymmetric reduction by a catalyst or enzyme or by hydride reduction," Magnus explains. "But an early experiment using aryl transfer chemistry resulted in very high conversions and very good enantioselectivity, and given the timeline we were up against, we went with that."
The synthesis starts with a postulated arylalkylzinc species, which is prepared by exposing a boroxine to diethylzinc. Reacting this with 3-cyanobenzaldehyde in the presence of an aminoalcohol organocatalyst yielded the desired diarylmethanol in greater than 94% ee. Reaction monitoring and analysis to understand optimal stoichiometries and reaction end points helped control the process efficiently.
In these reactions, a polymer additive often improves selectivity. Removing it meant the catalyst loading had to be increased slightly to maintain selectivity, but it allowed for an easier and cleaner workup and catalyst recycling. Ultimately, Magnus believes the most cost-effective, long-term solution would be an asymmetric reduction of a ketone by catalytic hydrogenation or an enzymatic process to give a chiral alcohol.
A GOAL of early-phase process development is to rapidly identify a stable advanced intermediate around which one can set preliminary purity specifications, Magnus explains. The technology for preparing this intermediate is either developed internally or by a third-party provider, which ultimately prepares the material in bulk.
This strategy enables more time for the internal process scientists to focus on developing the late-stage synthetic steps to ensure control of impurities in the final product. In a company's own plants or at an outside provider, an advanced intermediate will undergo its final transformations under strict regulatory conditions to become a product.
Another aspect of process development, mentioned by almost all pharmaceutical company process chemists who spoke with C&EN, is the need for determining an "E factor" for a process. "A health, safety, and environmental assessment is a critical criterion for acceptability in our process development," Magnus says. "It's got to be dealt with, and it's not taken lightly. If problems are insurmountable, or there is danger to the environment, a synthesis will be replaced."
Cover Story
- Centering On Chirality
- Chemists are finding asymmetric synthesis increasingly handy for making pharmaceutical compounds at large scale
- Web Exclusive - New Horizons Along Asymmetric Routes
- Academic researchers describe new synthetic approaches at Chiral Chemistry meeting
Cover Story
- Centering On Chirality
- Chemists are finding asymmetric synthesis increasingly handy for making pharmaceutical compounds at large scale
- Web Exclusive - New Horizons Along Asymmetric Routes
- Academic researchers describe new synthetic approaches at Chiral Chemistry meeting
Related Stories
- » Chirality From Counterions
- C&EN, July 30, 2007
- » A Barely Chiral Molecule, Exposed
- C&EN, April 02, 2007
- » Creating Chirality
- C&EN, September 11, 2006
- » Removing Impurities
- C&EN, September 5, 2005
- ?? Chiral Catalysis
- C&EN, September 5, 2005
- » Enzymes At Work
- C&EN, August 14, 2006
- » Market Challenges, Patients And Activists, And Industry Consolidation: 1980 - Present
- C&EN, June 20, 2005