-
September 14, 2009 - Volume 87, Number 37
- pp. 11 - 16
Cover Story - Science & Technology
 View Enlarged Image
View Enlarged Image
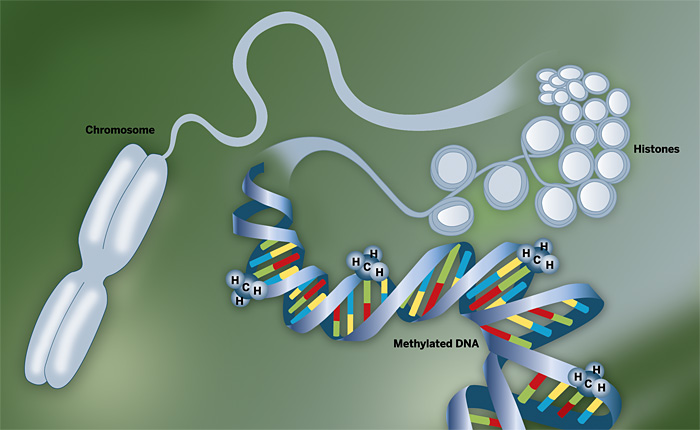
UNWRAPPED Common epigenetic modifications include methylation of genomic DNA and acetylation of the histone proteins that package genomic DNA into chromatin.
Related Stories
- Orchestrating Genetic Expression
- Another Epigenetics Agreement Signed
- Key Advance In DNA Sequencing With Nanopores
Topics Covered

METHYL MAPPING Sodium bisulfite treatment converts unmethylated cytosine residues (green) in genomic DNA to uracil (blue). Methylated cytosines (red) resist conversion and are identified by microarrays or sequencing. Gold blocks indicate common adapter DNA sequences ligated to the ends of each DNA fragment to enable amplification by polymerase chain reaction (PCR).
 View Enlarged Image
Brock Christensen/Adapted from PLoS Genetics 2009, 5, e1000602
View Enlarged Image
Brock Christensen/Adapted from PLoS Genetics 2009, 5, e1000602
TISSUE MATTERS Microarray analysis of bisulfite-converted DNA reveals tissue-specific DNA methylation patterns.
Each row on the microarray image is a
CpG dinucleotide, and each column is a tissue sample from a different person. Blocks of color above columns indicate tissue sample type.
 Karolin Luger/Colorado State U
Karolin Luger/Colorado State U
MARKED In this crystal structure of a nucleosome, selected sites of acetylation (green), methylation (gold), methylation or acetylation (red), and phosphorylation (blue) on various histones (white) are indicated. The DNA duplex is shown in amber.

If the human genome is the “Book of Life,” the epigenome is an anthology of closely related yet distinct tomes. The sequence of A, T, G, and C nucleotides in the genome remains constant throughout a person’s lifetime, but chemical modifications of DNA and its packaging proteins, known as histones, vary with tissue type, development, environmental conditions, aging, and cancer. These epigenetic modifications, which include DNA cytosine methylation and addition of various chemical groups to histones, cause dramatic changes in gene expression without altering the underlying DNA sequence. Each of us has one genome but many epigenomes, and that makes the Human Genome Project seem like a walk in the park compared with the marathon task of genomewide epigenetic mapping.
Growing recognition of the importance of epigenetic processes in human development and disease has fueled an insatiable thirst for new technologies to detect epigenetic modifications on a genomewide, or “epigenomic,” scale. The complex, dynamic nature of epigenetic modifications places many demands on analytical tools, but these challenges haven’t stopped researchers from developing powerful techniques for epigenomic analysis. With existing methods and others on the horizon, scientists hope to reach beyond the genome to understand how small chemical groups can orchestrate big changes in gene expression.
“This is a crazily innovative field, both in the research that’s being done and on the technology front,” says Pete Jozsi, editor of EpiGenie, a website that provides epigenetics-related news and technology information to the research community.
Peter W. Laird, associate professor of biochemistry and molecular biology and epigenetics researcher at the University of Southern California, comments: “One of the challenges for everyone in this field is writing grants, because it’s a moving target. What technology do you propose, because by the end of the grant you know that you’re going to be doing something completely different from what you’re doing now.”
The already fast-paced field received an additional boost from the National Institutes of Health Roadmap Epigenomics Program, launched in 2008. The program, which will invest more than $190 million in funded projects over the next five years, supports the development of new technologies for mapping epigenetic marks and establishes reference epigenome mapping centers. “The scientists we talked to suggested that there were some really important gaps in what we are able to do in epigenomics,” says John Satterlee, a program director for the initiative at the National Institute on Drug Abuse. “We asked investigators to come up with revolutionary technologies that have the potential to significantly change the way epigenomics research is performed in the future.”
“The aims of the Roadmap Epigenomics Program address many of the issues that plagued other areas of research in their early days, like a lack of standardized analytical methods,” Jozsi remarks. “It’s pretty tough to collaborate and compare data sets when you have hundreds of active research groups using at least 10 different approaches.” This heterogeneity of methods is particularly prevalent in DNA methylation research.
In plants and animals, DNA methylation, which is catalyzed by methyltransferase enzymes, occurs at the C-5 position of cytosine residues that precede guanines (so-called CpG dinucleotides). Long stretches of CpG-rich DNA frequently reside near promoter regions of genes, and abundant methylation of these “CpG islands” can silence genes. The differences between the transcriptional effects of cytosine and 5-methylcytosine are so dramatic that the latter is sometimes referred to as “the fifth base.” Nevertheless, the two have equivalent base-pairing properties and can’t be distinguished by standard DNA sequencing or microarray hybridization.
To map methylcytosines in DNA, the gold-standard approach involves treatment of DNA with sodium bisulfite, which converts unmethylated cytosine to uracil while leaving methylated cytosine intact (Nucleic Acids Res. 1994, 22, 2990). This technique revolutionized DNA methylation research by preserving DNA methylation status in a manner that can be amplified, cloned, and sequenced.
After bisulfite conversion, many researchers detect methylated DNA by hybridization to high-density microarrays. Brock Christensen and Karl Kelsey of Brown University and coworkers recently adopted this approach to investigate human DNA methylation changes in response to environmental exposures (PLoS Genetics 2009, 5, e1000602).
“Most previous studies of this type have involved either very few people or very few loci, which doesn’t truly assess epigenome variability,” explains Kelsey, a professor of community health and pathology and laboratory medicine. Therefore, in a large-scale study, the researchers treated DNA from more than 200 normal human tissue samples with sodium bisulfite. Then, they identified methylated loci by hybridizing the DNA to a microarray that contained probes for more than 1,500 CpG sites from hundreds of genes. Their results revealed striking variations in DNA methylation patterns with tissue type and with environmental exposures such as smoking.
Although the relative ease and cost-effectiveness of microarray-based readouts have made this technology the workhorse of DNA methylation research in recent years, scientists are limited to analyzing the CpG sites included in the array. These CpG sites represent only a tiny fraction of those found in the genome. Increasingly, researchers are turning to high-throughput sequencing to analyze bisulfite-converted DNA in a less biased manner. In this powerful technique, tens of millions of short DNA fragments are bound to the surface of a flow cell and sequenced in parallel. Then, researchers computationally map the short sequence reads to a reference genome, and in the case of bisulfite sequencing, identify the methylated cytosines (in other words, those not converted to uracil). Because individual DNA fragments from thousands of cells are sequenced in parallel, high-throughput sequencing enables absolute quantitation of the methylation level at a particular CpG site, an important characteristic for epigenomic studies.
The combination of bisulfite conversion with high-throughput sequencing has allowed researchers to achieve the monumental task of mapping every methylated cytosine in the genome of the plant Arabidopsis thaliana (Nature 2008, 452, 215; Cell 2008, 133, 523). But for comprehensive methylation mapping of the much larger human genome, the cost of bisulfite sequencing is prohibitive, especially because many samples must be sequenced to study tissue- and disease-specific variability.
Several researchers, however, have devised methods to reduce the epigenome to a manageable size for high-throughput sequencing. For example, Kun Zhang, an assistant professor of bioengineering at the University of California, San Diego, and coworkers recently designed a set of approximately 30,000 “padlock probes” to capture, in a single test tube, around 66,000 CpG sites on three different human chromosomes (Nat. Biotechnol. 2009, 27, 353). Each padlock probe consists of a common linker nucleotide sequence flanked by two target-specific arms, which enable annealing to and duplication of bisulfite-treated DNA at the targeted CpG site. The captured DNA is then subjected to high-throughput sequencing. George M. Church of Harvard University and colleagues independently developed a similar method (Nat. Biotechnol. 2009, 27, 361).
When the researchers examined the expression of genes associated with the analyzed CpG islands, they found that active genes had unmethylated promoters but methylated coding regions. This finding contradicts the conventional wisdom that DNA methylation always silences genes and demonstrates the importance of analyzing CpG sites outside promoter regions. Now Zhang, the recipient of an NIH Roadmap Epigenomics Technology Development grant, is scaling up the method with the goal of analyzing the methylation status of most CpG sites in the human genome.
Using a different approach, James Hicks and Gregory J. Hannon, professors of genetics at Cold Spring Harbor Laboratory, and coworkers profiled more than 25,000 CpG sites throughout the human genome (Genome Res., DOI: 10.1101/gr.095190.109). To isolate the selected CpG sites, the team designed a custom “capture array” that contained probes for methylated and unmethylated versions of the target sites. Fragmented, bisulfite-treated DNA from normal or cancer cells was hybridized to the array, and captured CpG sites were eluted and analyzed by high-throughput sequencing.
Again, the ability to profile thousands of CpG sites at single-nucleotide resolution provided new insights into the complexities of DNA methylation patterns. “One of the biggest surprises from this analysis is that about 10% of the CpG islands have subregions that can be methylated independently of one another,” Hicks says. The investigators noticed that these sharp transitions in methylation status often overlap important genetic elements such as transcription start sites.
Methods such as Zhang’s and Hicks’s are allowing scientists to study DNA methylation across the human genome in unprecedented detail, but the captured CpG sites still represent less than 0.1% of the human genome. “Right now, it’s more cost-effective to limit yourself to subsets of the genome,” Laird says. “The caveat is that you’re looking under the streetlamp, so you won’t identify things in regions of the genome that haven’t been well investigated.”
Because the cost of high-throughput sequencing continues to decline, many researchers believe that whole-genome bisulfite sequencing will soon replace microarray readouts and targeted bisulfite sequencing for analysis of human DNA methylation. “The Human Genome Project took 12 years and $3 billion to complete,” Laird says. “Then, last year, the entire human genome was resequenced with high-throughput sequencing for a few hundred thousand dollars, and this year we’re down to $50,000. I would be very surprised if we didn’t reach the $1,000 mark in the next five years.”
Less costly high-throughput sequencing will likewise be a boon for another important area of epigenetic research: the analysis of histone modifications. Histones are spool-like proteins that compact about 6 feet of linear DNA into an approximately 10-μm cell nucleus. Two molecules each of the histone proteins H2A, H2B, H3, and H4 form an octameric complex, around which a 147-base-pair length of DNA wraps to form a “nucleosome.” “Chromatin,” a complex tapestry of nucleosomes and other proteins, folds into a series of higher order structures to ultimately package DNA into chromosomes. Changes in chromatin structure affect the accessibility of DNA to various proteins.
Histones are subject to more than 100 posttranslational modifications, including methylation, acetylation, and phosphorylation. Histone modifications, which occur primarily in the flexible N-terminal “tail” regions of the proteins, affect the likelihood that bound DNA will be transcribed. Certain modifications, such as lysine acetylation, can directly loosen chromatin structure to increase DNA accessibility to transcriptional complexes, whereas other modifications recruit specific transcriptional activators or repressors to turn gene expression on or off.
To identify locations of modified histones throughout the genome, researchers precipitate cross-linked, fragmented chromatin with an antibody that recognizes a specific modification—a procedure known as chromatin immunoprecipitation. The DNA sequences associated with that modified histone are released and then identified by microarray hyridization or high-throughput DNA sequencing.
As in the DNA methylation field, many researchers who study histone modifications are embracing high-throughput DNA sequencing technologies. Bradley Bernstein, an associate professor of pathology at Harvard, and colleagues used chromatin immunoprecipitation combined with high-throughput sequencing to map the locations of five histone methyl marks in the genomes of mouse cells at different stages of differentiation (Nature 2007, 448, 553). The team correlated these histone modifications with gene expression data. “The data reveal the power of chromatin modification maps to identify and characterize genome regulatory elements” such as promoters, Bernstein says.
Because Bernstein’s lab has been designated as an NIH Roadmap Reference Epigenome Mapping Center, Bernstein is now gearing up for the ambitious project of mapping both chromatin and DNA methylation states in the genomes of 100 human cell types. “The mapping centers will provide a framework of ‘normal’ epigenomic maps for various tissues, which could then be used to identify epigenomic differences in diseases,” Bernstein says.
Histone modification is a dynamic process. Not only do enzymes modify histones bound to DNA, but nucleosomes are also thought to assemble and disassemble at different points in the cell cycle and in the process swap old histones with new ones that harbor various posttranslational modifications. However, current methods provide only a snapshot of the average modification state of a particular nucleosome in a population of cells. “Right now, when we say there’s a combination of posttranslational modifications in a given nucleosome, we don’t even know that they’re there at the same time,” says Steven Henikoff of the basic sciences division at the Fred Hutchinson Cancer Research Center.
To address this problem, Henikoff’s lab is developing a potentially groundbreaking technique called CATCH-IT (Covalent Attachment of Tags to Capture Histones and Identify Turnover), with the support of NIH Roadmap Epigenomics funding. Using CATCH-IT, Henikoff plans to study histone replacement dynamics throughout the genome. If successful, the method would allow researchers to isolate newly synthesized histones from cells at various times so they can observe how quickly histones (and histone modifications) are replaced at particular sites in the genome.
In the CATCH-IT technique, Henikoff’s group metabolically labels all newly synthesized proteins in cells with the methionine analog azidohomoalanine. They then isolate chromatin and cleave it into single-nucleosome fragments by using an enzyme. With a copper catalyst, the researchers chemically attach a biotin moiety to the azide group in labeled histones. Using streptavidin, a protein that binds strongly to biotin, they then isolate nucleosomes containing newly synthesized histones. The team is analyzing data from preliminary experiments. “At present, we’ve achieved proof of concept and have applied CATCH-IT genome- wide to Drosophila cells,” Henikoff says.
Cornell University professors Paul D. Soloway and Harold G. Craighead are developing another potentially revolutionary technology with funding from the NIH Roadmap Epigenomics Program. Soloway, a professor of nutritional sciences, and Craighead, a professor of applied and engineering physics, are working on a nanoscale device to simultaneously detect independent epigenetic marks by fluorescence imaging. For this technique, the researchers first label specific modifications with affinity reagents that are conjugated to spectrally distinct fluorophores. Then, they flow the fluorescently labeled chromatin fragments through a channel of the nanoscale device at a rate of 4,000 molecules per minute. When a laser of the appropriate excitation wavelength impinges on an individual chromatin fragment, the characteristic fluorescence emission allows researchers to detect the modifications on that fragment.
“As chromatin flows through the channel and we image its fluorescence signature, we will be able to make a decision on the fly as to which chamber we want to shunt it into so that the material can be collected for downstream analysis,” Soloway says. For example, to correlate DNA methylation state with histone modifications, the researchers could sort chromatin fragments with specific histone marks into a chamber and perform high-throughput bisulfite sequencing of the associated DNA.
If successful, the nanoscale technology would greatly reduce sample requirements for epigenetic analysis. “Since we’re looking at individual molecules, we can use vanishingly small inputs of material,” Soloway says. “Even materials that can’t be cultured, for example, laser-microdissected material or the inner cell mass of a blastocyst, could be analyzed on our device.”
Innovative tools and improvements to existing technologies will offer breathtaking views of the epigenetic landscape across the entire genome. Nevertheless, the road to the epigenome will likely have bumps ahead—this field is full of surprises. As a case in point, two groups recently discovered an unexpected type of epigenetic modification. Working independently, teams at Harvard and Rockefeller University detected by chemical analysis significant amounts of 5-(hydroxymethyl)cytosine in mammalian DNA (Science 2009, 324, 929 and 930).
The biological function of 5-(hydroxymethyl)cytosine is unclear, but the scientists think it might recruit chromatin-remodeling proteins or serve as an intermediate for the oxidative demethylation of cytosine. In any case, researchers are scrambling to identify antibodies or other reagents that would allow large-scale discrimination between the two cytosine modifications, which are similarly resistant to bisulfite conversion and therefore cannot be distinguished by bisulfite sequencing. Many researchers believe that additional DNA and histone modifications remain to be discovered.
With a brave new world awaiting exploration beyond the genome, it’s not surprising that epigenetics has risen from near obscurity to become one of the hottest fields in biology. By probing the multifaceted human epigenome in a variety of cell types, researchers will gain a better understanding of both normal and pathological epigenetic states. “I hope that by the end of the Roadmap Program, epigenomic analysis won’t be something that only experts utilize,” Satterlee says. “Rather, it will be used routinely in labs to investigate disease and develop therapies.”
- Chemical & Engineering News
- ISSN 0009-2347
- Copyright © 2011 American Chemical Society
Services & Tools
ACS Resources
ACS Careers
ACS is the leading employment source for recruiting scientific professionals. ACS Careers and C&EN Classifieds provide employers direct access to scientific talent both in print and online. Jobseekers | Employers
» Join ACS
Join more than 161,000 professionals in the chemical sciences world-wide, as a member of the American Chemical Society.
» Join Now!
Popular Sections
ACS, Analytical SCENE, Business, Careers, Economy, Editor, Education, Employment, Environmental SCENE, Letters, Government, Photo Galleries, Policy, Movies, Multimedia, Nanotechnology, Newscripts, Reel Science, Safety Letters, Science, Stem Cells, Stimulus Funds, Technology, Vaccines, Videos, What's That Stuff
Advertisements
