Advertisement
Advertise Here
-
September 05, 2011 - Volume 89, Number 36
- pp. 19 - 26
 Juswinder Singh and Aravind Prasad/Avila Therapeutics
Juswinder Singh and Aravind Prasad/Avila Therapeutics
Forensic Chemistry: A new method could increase the number of explosives detected by airport screeners.
Trade: U.S. companies complain of market dumping by China.
Layoffs follow similar moves by Amgen, AstraZeneca.
Environment: Ban to halt export of hazardous waste to developing world.
Penrose (Parney) Albright will direct DOE national lab.
Toxic Exposure: Mercury isotopes in human hair illuminate dietary and industrial sources.
Cancer Biochemistry: Mass spectrometry follows the metabolism of very long fatty acids in cancer cells.
When most small-molecule drugs meet a protein target, they nestle close to their intended. But the flirtatious relationship between drug and target is often fleeting, with the drug repeatedly drifting away and coming back. Like a teenager with no interest in settling down, the drug forms no permanent bond with its target.
Some small molecules, however, do forge lasting ties. Their irreversible covalent bonds with proteins link compound and protein for good.
The pharmaceutical industry has by and large avoided developing drugs interested in marriage and instead has pursued dating as a model of the drug-target interaction. Covalent drugs, companies have feared, would be so desperate for a mate that they would bond with the wrong proteins. The result would undoubtedly be toxicity.
But now a growing group of researchers, in academe and in companies large and small, has begun pursuing covalent drug candidates. They say the strategy could provide high potency and selectivity—along with low toxicity. They aim to treat a variety of conditions including cancer, hepatitis C, and obesity.
And they’re changing minds that were once reluctant to consider marriage.
Several drugs designed to form covalent bonds with their targets are in large-scale clinical trials and could hit the market within a few years. Still more are in preclinical or early-stage clinical research.
“There are pharmacological problems that covalent drugs are uniquely positioned to solve,” says Juswinder Singh, founder and chief scientific officer of Avila Therapeutics, a small company in Bedford, Mass., that champions the discovery and development of covalent drugs. “Those results would be nearly impossible with reversible inhibitors.”
Covalent drugs that form irreversible chemical bonds with their protein targets provide many advantages, according to their developers. (By contrast, covalent inhibitors that form reversible bonds—those that repeatedly marry, divorce, and remarry—behave more like noncovalent drugs, particularly if they divorce quickly.) One strength stems from the drugs’ very nature: The marriage between drug and target permanently prevents the protein molecule from wreaking havoc.
As a result, patients may need to receive doses that are only high enough for one drug molecule to reach each target protein molecule. And patients shouldn’t need a second dose until their body has made more protein, often a day or more later. They end up with very low concentrations of the molecule in their bloodstream.
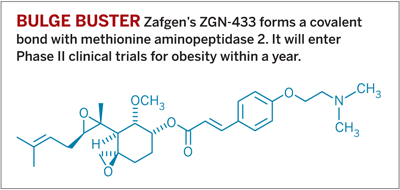
At such low doses, says Thomas E. Hughes, president and chief executive officer of Zafgen, toxicity concerns tend to evaporate, in part because so little opportunity exists to inhibit off-target proteins. Zafgen, a small pharmaceutical company in Cambridge, Mass., sees high selectivity and low toxicity with its covalent molecule for treating obesity, beloranib hemioxalate, also known as ZGN-433.
“You’re passing a wave of the molecule through the body,” he says. “It hits the different tissues, silences the target enzyme where it finds it, and then it goes away.”
Zafgen’s drug candidate inhibits an enzyme called methionine aminopeptidase 2 (MetAP2), which had been of interest in oncology circles until it turned out to be a poor target for treating cancer in mice. However, animals treated with a MetAP2 inhibitor lost weight. Zafgen pursued the enzyme as a target for obesity.
Its drug candidate contains a spiroepoxide that bonds with a histidine in the protein’s active site. ZGN-433 has undergone a Phase I clinical trial, in which obese volunteers lost up to 2 lb per week. It will enter Phase II trials within a year, Hughes says, funded by $33 million the company raised from investors.
With dosing of up to 2 mg twice per week, ZGN-433 reaches a maximum concentration in the body of just a few nanomolar for several hours before the body quickly eliminates it, Hughes says. During that time, the drug is much more likely to interact with MetAP2 than with anything else.
“You’re flying under the radar of a lot of concerns,” he says. “Drug-drug interactions are not an issue. There’s just not enough inhibitor to go around. The same is true for off-target inhibition: The chance of off-target toxicity is largely gone.”
Proponents of covalent inhibitors are quick to point out that dozens of such drugs are already on the market. They include aspirin, the world’s most widely used medicine; penicillin and related antibiotics; and recently developed blockbusters such as Plavix, Prevacid, and Nexium. The drugs treat a broad range of conditions, and many have minimal side effects, even when taken for years. By one count, of the marketed drugs that inhibit enzymes, more than one-third work by covalent modification (Biochemistry, DOI: 10.1021/bi050247e).
“Nobody can argue with the historical success of covalent drugs,” Singh says.
But irreversible inhibitors on the market are not covalent by design. Their chemical mechanisms came to light after the molecules were designed or discovered, sometimes years after they hit the market.
Drug companies have tried to avoid such permanent connections. As in many human relationships, the reason for companies’ avoiding the ties that bind has been fear. Companies were afraid of toxicity stemming from the permanent nature of the bond between the drug and protein.
Small molecules that are reactive enough to covalently modify their targets, the thinking goes, could be promiscuous and react with other proteins, says Thomas A. Baillie, dean of the School of Pharmacy at the University of Washington, Seattle. Baillie, who spent 14 years at Merck & Co., is an expert on toxicity associated with reactive drug metabolites—and thinks that those very reactive, very electrophilic metabolites gave covalent drugs a bad reputation.
“But there’s a fundamental difference,” he says: The new covalent drugs are only weakly reactive and are designed to be highly selective.
Still, even a covalent modifier that bonds only with its target may ring companies’ alarm bells because of concern about so-called idiosyncratic toxicity. The immune system causes these unpredictable, rare, often serious side effects such as liver failure or blood disorders. The rarity of the toxicity means that it may not rear its head until the drug is in large-scale clinical trials or even on the market.
The results can be fatal—for the patient and for the drug or drug candidate.
“The worst thing that can happen to a firm is that it spends a lot of money developing a compound, it gets registered and gets on the market, and then you find out there are idiosyncratic life-threatening events,” says Mark E. Duggan, a veteran of Amgen who now is vice president of chemistry at Link Medicine in Cambridge, Mass. “That can cause the drug to be withdrawn from the market. That’s a huge hit that the company takes.”
But Duggan and others point out that no systematic study has proven that covalent bonding leads to idiosyncratic effects.
“The evidence is largely circumstantial,” Baillie says. Some animal studies have found a correlation between the extent of irreversible binding and the severity of tissue damage. “Much remains to be learned about the factors that dictate the toxicity, or lack thereof, of reactive electrophiles,” he says.
As with tales of failed marriages, anecdotes carry weight. Like many others who have worked in pharma, Christopher A. Lipinski can tell of horrifying idiosyncratic toxicity from covalent drug candidates. Lipinski is retired from Pfizer and is famous for creating guidelines to reduce risk in developing new drugs.
He recalls Pfizer’s experience with sorbinil, an aldose reductase inhibitor that the company tested in the 1980s to treat complications of diabetes. It gave some people a mild skin rash. For others, though, the outcome was far worse: “Literally, your skin peeled off,” Lipinski says. “People ended up in burn wards.” After a Phase III study, the company stopped working on the drug candidate—and on other molecules that contained the same reactive heterocycle, a spiro hydantoin.
Lipinski argues that, with the possible exception of drugs for life-threatening diseases, pharmaceutical companies should continue to stay away from covalent drugs. “Should we move forward with a flawed compound that we can identify early on?” he asks. The answer, Lipinski says, is no. When asked about Zafgen’s drug candidate for obesity, he takes one look at the structure and laughs. “I wish them luck,” he says.
Others defend Zafgen’s strategy. When it comes to covalent drugs, Singh says, many researches like Lipinski “have underestimated the benefits and overestimated the risks. They’ve given up on a whole therapeutic class.” Besides, Singh says, sorbinil is not a covalent drug. Instead it is a noncovalent, reversible drug that may be metabolized to form a reactive covalent intermediate. “It’s important,” he says, “that we don’t confuse selective covalent drugs with indiscriminate covalent metabolites.”

Chemistry and chemical biology professor Jack Taunton of the University of California, San Francisco, also encourages the covalent drug approach. He notes, “Many drugs form irreversible covalent bonds but have great safety records.”
A potential covalent drug success story stems from work begun in 1994, when David W. Fry and colleagues at Parke-Davis discovered a surprisingly potent reversible inhibitor of epidermal growth factor receptor (EGFR) tyrosine kinase, an enzyme overexpressed in several types of cancer (Science, DOI: 10.1126/science.8066447). Next they wanted to suppress the target for long periods, to increase the inhibitor’s in vivo antitumor activity, says Fry, now the chief scientific officer of Everist Genomics, in Ann Arbor, Mich. “What better way than to irreversibly knock the receptor out?” he asks.
Only one crystal structure of a kinase was available at the time, so Singh, a computational chemist who was then Fry’s colleague at Parke-Davis, built a homology model to study how natural substrates bind to EGFR. The researchers began assembling molecules based on their inhibitor, which binds reversibly to the adenosine triphosphate (ATP) pocket of EGFR. They added alkylating groups to the structure, to form a covalent bond via a Michael addition with a cysteine that Singh identified near the active site. Such a strategy, he notes, would help the drug be more selective for the target, despite the high concentrations of ATP in cells.

Convincing others in the company to move forward with an irreversible inhibitor was the next challenge. “There was a lot of skepticism,” Singh recalls. But the compound’s preclinical success spoke for itself. Its toxicity was unremarkable and its antitumor activity was “pretty spectacular,” Fry says.
When Pfizer purchased Parke-Davis in 2000, Fry continued work on the irreversible inhibitor. The drug candidate went into the clinic, Fry says, “and lo and behold, it didn’t kill people.”
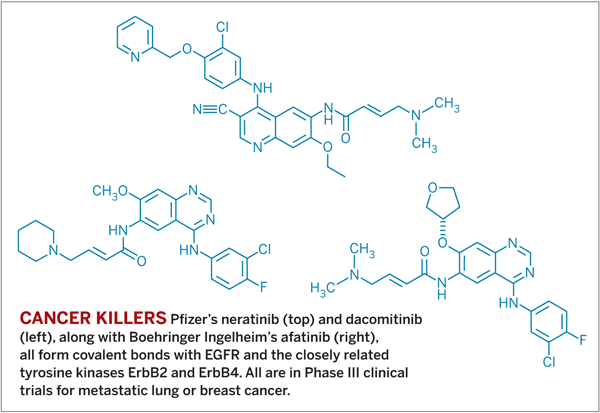
The company stopped work on the drug candidate after Phase II studies, but it did move forward with two other irreversible inhibitors of EGFR. Each of them is in Phase III clinical trials, and they have been taken by hundreds of people with metastatic lung or breast cancer. Their side effects, including diarrhea, are similar to those of conventional reversible inhibitors.
“Patients stay on the therapy and have a low rate of discontinuation,” says Joseph O’Connell, a medical oncologist at Pfizer who leads clinical work on one of the compounds. Known as dacomitinib, it is a compound that Fry’s team originally developed.
Pfizer’s second candidate, neratinib, was originally developed at Wyeth. That company started work on the irreversible drug after reading Fry’s Science paper.
In the early stages of research on the drug, “it wasn’t clear if you could design a selective inhibitor of a tyrosine kinase,” recalls Allan Wissner, who retired from Pfizer last year. Because tyrosine kinases have very similar active sites, one small molecule might inhibit them all. Nevertheless, he says, “we did know that the cysteine we targeted in EGFR was relatively rare. It only existed in 11 other kinases.”
Both of the Pfizer irreversible inhibitors have since been found to selectively target EGFR and two closely related tyrosine kinases, ErbB2, which is better known as HER2, and ErbB4. They do so without inhibiting the hundreds of other tyrosine kinases in the human body.
Both are also effective in the clinic against tumors that have developed resistance to the reversible inhibitors Iressa and Tarceva. The drug-resistant forms of EGFR have a misshapen active site but can still covalently bond to the irreversible inhibitors.
“As long as the covalent drug is on the target long enough to form a bond, you overcome the resistance,” Singh says. “You’re able to deal with some very problematic mutations.” Some companies, including Avila, are now working to develop covalent drugs that target only the mutated forms of the enzyme.
Boehringer Ingelheim also has developed an EGFR-family inhibitor that is in Phase III trials. More than 1,800 patients have taken the experimental drug, called afatinib.
Other anticancer covalent drugs in clinical development include a molecule from Onyx Pharmaceuticals that targets the proteasome and a Sanofi drug candidate that targets the enzyme PARP1. Pharmacyclics, of Sunnyvale, Calif., and Avila are each testing a covalent inhibitor of Bruton’s tyrosine kinase as treatments for B-cell cancers, such as chronic lymphocytic leukemia and mantle cell lymphoma.
And Johnson & Johnson’s prostate cancer agent Zytiga forms a covalent bond with a cytochrome P450 enzyme. It received Food & Drug Administration approval in April.
Although some covalent drugs react directly with an enzyme’s catalytic site, many drugs under development take the same tack as the EGFR inhibitors: They form a covalent bond with a noncatalytic amino acid that is unique to the target. A covalent drug designed this way might interact noncovalently with related, off-target structures or proteins, but it will form a tight, permanent bond only with its target (Nat. Rev. Drug Discov., DOI: 10.1038/nrd3410).
Knowledge of three-dimensional target structures allows Avila and other companies to design irreversible inhibitors that bind reversibly to protein active sites, providing time for nearby chemical reactions that permanently bond drugs to targets. The reversible binding makes the drug’s concentration at the target effectively sky-high, helping drive the chemical reaction of a nucleophilic amino acid on the protein with a modestly electrophilic moiety on the drug.
The strategy works with proteins in general. Avila, for example, is doing preclinical testing of a covalent inhibitor of a noncatalytic structure on hepatitis C virus protease (C&EN Online Latest News, Dec. 1, 2010). And chemist Benjamin F. Cravatt of Scripps Research Institute designed an inhibitor of fatty acid amide hydrolase as a possible pain treatment (ACS Med. Chem. Lett., DOI: 10.1021/ml100190t). It covalently bonds with a noncatalytic serine on the enzyme. Pfizer did Phase I and II clinical trials on the molecule but stopped work on it last year.
“This approach should be applicable to any target class,” says UCSF’s Taunton, who has worked on irreversible kinase inhibitors. Aside from kinase inhibitors, there aren’t many examples of covalent drugs that were designed to target noncatalytic amino acids. But, he says, such approaches are “really powerful” and could be used more broadly.
However, the strategy won’t work for just any target molecule. For example, to bond with an electrophilic drug, a protein should have a nonconserved nucleophilic amino acid near a small-molecule binding site, and not all do.
Designing covalent inhibitor drugs always requires great care. Some drug targets should not be shut down permanently. If drugs that prevent blood clots turn off coagulation completely, patients could suffer from excess bleeding. “There is a very fine balance between too much coagulation and not enough coagulation,” comments Koen Augustyns, a professor of medicinal chemistry at the University of Antwerp, in Belgium.
For many proteins, targeted covalent drugs may make perfect mates, but that’s challenging to prove. “There is reason to believe that there is a lower likelihood of their being toxic, but that has to be demonstrated,” says Baillie, who serves on Avila’s scientific advisory board. “The jury is still out as to whether this approach will be associated with a better safety profile.”
Covalent drugs’ proponents see their ranks as small but growing, as they turn others on to the benefits of irreversible ties. But they acknowledge that skepticism remains.
“You’re never going to be able to convince everybody,” Singh says.
It’s true in developing pharmaceuticals, as in life: Not everyone is the marrying type.
ACS is the leading employment source for recruiting scientific professionals. ACS Careers and C&EN Classifieds provide employers direct access to scientific talent both in print and online. Jobseekers | Employers
Join more than 161,000 professionals in the chemical sciences world-wide, as a member of the American Chemical Society.
» Join Now!